Unit 5 Thermodynamics
5.6 Spontaneity of a Chemical Reaction
OpenStax
Section Learning Objectives
- Evaluate Gibbs (free) energy changes for chemical processes.
- Predict the spontaneity of a chemical reaction.
- Describe how spontaneity varies with temperature.
✓ SECTION 5.6 CHECKLIST
| Learning Activity | Graded? | Estimated Time |
|---|---|---|
| Make notes about this section’s reading portion. | No | 60 min |
| Optional Activity: Perform a virtual lab about the spontaneity of phase changes. | No | 20 min |
| Work on the self-check question. | No | 10 min |
| Work on practice exercises. | No | 200 min |
| Complete Unit 5 Assignment on Moodle. | Yes | 200 min |
📖 READING PORTION
Gibbs Free Energy
One of the challenges of using the Second Law of Thermodynamics to determine if a process is spontaneous is that we must determine the entropy change for the system and the entropy change for the surroundings. An alternative approach involving a new thermodynamic property defined in terms of system properties only was introduced in the late nineteenth century by American mathematician Josiah Willard Gibbs. This new property is called the Gibbs free energy (G) (or simply the free energy), and it is defined in terms of a system’s enthalpy and entropy as
[latex]G = H\;-\;TS[/latex]
Free energy is a state function, and at constant temperature and pressure, the free energy change (ΔG) may be expressed as
Equation 1
[latex]{\Delta}G = {\Delta}H\;-\;T{\Delta}S[/latex]
- [latex]\Delta G[/latex], change in Gibbs free energy ([latex]\text{J}[/latex] or [latex]\text{J}/\text{mol}[/latex])
- [latex]\Delta H[/latex], change in enthalpy ([latex]\text{J}[/latex] or [latex]\text{J}/\text{mol}[/latex])
- [latex]T[/latex], kelvin or absolute temperature
- [latex]\Delta S[/latex], change in entropy ([latex]\frac{\text{J}}{\text{K}}[/latex] or [latex]\frac{\text{J}}{\text{mol}\cdot \text{K}}[/latex])
The choice to include the unit of mol for ΔG, ΔH, and ΔS depends on what the question is asking for. Whether or not we include mol, we need to be consistent for all three quantities ΔG, ΔH, and ΔS. When the unit of mol is included, denoting a molar property, we tend to use the notation ΔGr, where the subscript “r” means “per mole of reaction”. Such notation is consistent with molar enthalpy change (ΔHr) and molar entropy change (ΔSr).
The standard change in Gibbs free energy (ΔG°) is calculated using standard enthalpy change (ΔH°) and standard entropy change (ΔS°).
We can understand the relationship between this system property and the spontaneity of a process by recalling the previously derived Second Law expression
[latex]{\Delta}S_{\text{univ}} = {\Delta}S\;+\;\frac{q_{\text{surr}}}{T}[/latex]
The First Law requires that qsurr = −qsys, and at constant pressure qsys = ΔH, and so this expression may be rewritten as
[latex]{\Delta}S_{\text{univ}} = {\Delta}S\;-\;\frac{{\Delta}H}{T}[/latex]
ΔH is the enthalpy change of the system. Multiplying both sides of this equation by −T, and rearranging yields
[latex]-T{\Delta}S_{\text{univ}} = {\Delta}H\;-\;T{\Delta}S[/latex]
Comparing this equation to the previous one for free energy change shows
[latex]{\Delta}G = -T{\Delta}S_{\text{univ}}[/latex]
The free energy change is therefore a reliable indicator of the spontaneity of a process, being directly related to the previously identified spontaneity indicator, ΔSuniv. Table 1 summarizes the relation between the spontaneity of a process and the arithmetic signs of these indicators.
|
ΔSuniv > 0 |
ΔG < 0 |
Spontaneous |
|
ΔSuniv < 0 |
ΔG > 0 |
Nonspontaneous |
|
ΔSuniv = 0 |
ΔG = 0 |
Reversible (at equilibrium) |
|
Table 1 Relation between Process Spontaneity and Signs of Thermodynamic Properties. Source: OpenStax Chemistry 2e |
||
Calculating Free Energy Change
Gibbs free energy is a state function, so its value depends only on the conditions of the initial and final states of the system that have undergone some change. A convenient and common approach to the calculation of free energy changes for physical and chemical reactions is by use of widely available compilations of standard state thermodynamic data. One method involves the use of standard enthalpies and entropies to compute standard free energy changes according to Equation 1.
The symbol ° on ΔG, ΔH, and ΔS refers to standard state, a special condition for the species involved in a reaction:
- the partial pressure of gases is 1 atm
- the concentration of dissolved solutes is 1 M
- gases and solutes behave ideally
- substances are in their most stable physical forms
Temperature is not part of the definition of standard state, but 298 K is a temperature at which most data are collected.
Example 1
Evaluation of ΔG°r from ΔH°r and ΔS°r
Use standard enthalpy and entropy data (Source: OpenStax Chemistry 2e) to calculate the molar standard free energy change for the vaporization of water at room temperature (298 K). What does the computed value for ΔG°vap say about the spontaneity of this process?
SOLUTION
The process of interest is
[latex]\text{H}_2\text{O}(l)\;{\longrightarrow}\;\text{H}_2\text{O}(g)[/latex]
The standard change in free energy may be calculated using Equation 1
[latex]{\Delta}G_{\text{vap, 298}}^{\circ} = {\Delta}H_{\text{vap, 298}}^{\circ}\;-\;T{\Delta}S_{\text{vap, 298}}^{\circ}[/latex]
The thermodynamic data at 298 K are
|
Substance |
[latex]{\Delta}H_{\text{f, 298}}^{\circ}(\text{kJ/mol})[/latex] |
[latex]S_{298}^{\circ}(\text{J/(K}{\cdot}\text{mol)})[/latex] |
|
H2O(l) |
−286.83 |
70.0 |
|
H2O(g) |
−241.82 |
188.8 |
(Source: OpenStax Chemistry 2e)
Combining at 298 K
[latex]{\Delta}H_{\text{vap, 298}}^{\circ} = {\Delta}H_{\text{f, 298}}^{\circ}(\text{H}_2\text{O}(g))\;-\;{\Delta}H_{\text{f, 298}}^{\circ}(\text{H}_2\text{O}(l)) = [-241.82\;-\;(-285.83)]\text{kJ/mol} = 44.01\;\text{kJ/mol}[/latex]
[latex]{\Delta}S_{\text{vap, 298}}^{\circ} = S_{298}^{\circ}(\text{H}_2\text{O}(g))\;-\;S_{298}^{\circ}(\text{H}_2\text{O}(l)) = (188.8\;\;-\;70.0)\text{J/(mol}\cdot\text{K)} = 118.8\;\text{J/(mol}{\cdot}\text{K)}[/latex]
Converting everything into kJ and combining at 298 K
[latex]\begin{array}{r @{{}={}} l} {\Delta}G_{\text{vap, 298}}^{\circ} &= {\Delta}H_{\text{vap, 298}}^{\circ}\;-\;T{\Delta}S_{\text{vap, 298}}^{\circ} \\[0.5em] &= 44.01\;\text{kJ/mol}\;-\;(298\;\text{K}\;\times\;118.8\;\text{J/(mol}{\cdot}\text{K)})\;\times\;\frac{1\;\text{kJ}}{1000\;\text{J}} \\[0.5em] &= 44.01\;\text{kJ/mol}\;-\;35.4\;\text{kJ/mol} = +8.6\;\text{kJ/mol} \end{array}[/latex]
At 298 K (25 °C) [latex]\boxed{{\Delta}G_{\text{vap, 298}}^{\circ}\;>\;0}[/latex], and so boiling is nonspontaneous (not spontaneous).
Free energy changes may also use the standard free energy of formation ([latex]{\Delta}G_{\text{f}}^{\circ}[/latex]), for each of the reactants and products involved in the reaction. The standard free energy of formation is the free energy change that accompanies the formation of one mole of a substance from its elements in their standard states. Similar to the standard enthalpies of formation, [latex]{\Delta}G_{\text{f}}^{\circ}[/latex] is by definition zero for elemental substances under standard state conditions. The approach to computing the free energy change for a reaction using this approach is the same as that demonstrated previously for enthalpy and entropy changes. At a given temperature and pressure,
Equation 2
[latex]{\Delta}G^\circ = {\sum}n\times {\Delta}G^\circ_\text{f}(\text{products})\;-\;{\sum}n\times {\Delta}G^\circ_\text{f} (\text{reactants})[/latex]
- [latex]\Delta G^\circ[/latex], standard Gibbs free energy change of reaction ([latex]\text{J}[/latex] or [latex]\text{J}/\text{mol}[/latex])
- [latex]n[/latex], stoichiometric coefficient ([latex]\text{mol}[/latex] or unitless)
- [latex]\Delta G^\circ_{\text{f}}[/latex], standard Gibbs free energy change of formation ([latex]\text{J}/\text{mol}[/latex])
The choice to include the unit of mol for ΔG° or stoichiometric coefficients depends on what the question is asking for. If stoichiometric coefficients have the unit mol, then ΔG° will be in J. If stoichiometric coefficients are unitless, then ΔG°r will have the unit J/mol.
For the reaction
[latex]a\text{A}\;+\;b\text{B}\;{\longrightarrow}\;c\text{C}\;+\;d\text{D}[/latex],
the standard free energy change at 298 K may be calculated as
[latex]{\Delta}G_{298}^{\circ} = {\Delta}G^{\circ} = {\sum}n\times {\Delta}G_{298}^{\circ}(\text{products})\;-\;{\sum}n\times {\Delta}G_{298}^{\circ}(\text{reactants}) = [c{\Delta}G_{\text{f, C}}^{\circ}\;+\;d{\Delta}G_{\text{f, D}}^{\circ}]\;-\;[a{\Delta}G_{\text{f, A}}^{\circ}\;+\;b{\Delta}G_{\text{f, B}}^{\circ}][/latex]
Example 2
Calculation of [latex]{\Delta}G_{298}^{\circ}[/latex]
Consider the decomposition of yellow mercury (II) oxide.
[latex]\text{HgO}(s\text{, yellow})\;{\longrightarrow}\;\text{Hg}(l)\;+\;\frac{1}{2}\text{O}_2(g)[/latex]
Calculate the standard free energy change at room temperature, [latex]{\Delta}G_{298}^{\circ}[/latex], using (a) standard enthalpies of formation and standard entropies (b) standard free energies of formation. Do the results indicate the reaction to be spontaneous or nonspontaneous under standard conditions?
SOLUTION
The thermodynamic data at 298 K are
|
Compound |
[latex]{\Delta}G_{\text{f}}^{\circ}(\text{kJ/mol})[/latex] |
[latex]{\Delta}H_{\text{f}}^{\circ}(\text{kJ/mol})[/latex] |
[latex]S^{\circ}(\text{J/(mol}\cdot\text{K)})[/latex] |
|
HgO (s, yellow) |
−58.43 |
−90.46 |
71.13 |
|
Hg(l) |
0 |
0 |
75.9 |
|
O2(g) |
0 |
0 |
205.2 |
|
|
|||
(Source: OpenStax Chemistry 2e)
(a) Using enthalpies and entropies of formation, Equation 1.
[latex]{\Delta}H_{298}^{\circ} = {\sum}n\times{\Delta}H_{298}^{\circ}(\text{products})\;-\;{\sum}n\times{\Delta}H_{298}^{\circ}(\text{reactants})[/latex]
[latex]= [1{\Delta}H_{298}^{\circ}\text{Hg}(l)\;+\;\frac{1}{2}{\Delta}H_{298}^{\circ}\text{O}_2(g)]\;-\;1{\Delta}H_{298}^{\circ}\text{HgO}(s\text{, yellow})[/latex]
[latex]= [1\;\text{mol}(0\;\text{kJ/mol})\;+\;\frac{1}{2}\text{mol}(0\;\text{kJ/mol})]\;-\;1\;\text{mol}(-90.46\;\text{kJ/mol}) = 90.46\;\text{kJ}[/latex]
[latex]{\Delta}S_{298}^{\circ} = {\sum}n\times{\Delta}S_{298}^{\circ}(\text{products})\;-\;{\sum}n\times{\Delta}S_{298}^{\circ}(\text{reactants})[/latex]
[latex]= [1{\Delta}S_{298}^{\circ}\text{Hg}(l)\;+\;\frac{1}{2}{\Delta}S_{298}^{\circ}\text{O}_2(g)]\;-\;1{\Delta}S_{298}^{\circ}\text{HgO}(s\text{, yellow})[/latex]
[latex]= [1\;\text{mol}(75.9\;\text{J/mol K})\;+\;\frac{1}{2}\text{mol}(205.2\;\text{J/mol K})]\;-\;1\;\text{mol}(71.13\;\text{J/mol K}) = 107.4\;\text{J/K}[/latex]
[latex]{\Delta}G^{\circ} = {\Delta}H^{\circ}\;-\;T{\Delta}S^{\circ} = 90.46\;\text{kJ}\;-\;298.15\;\text{K}\;\times\;107.4\;\text{J/K}\;\times\;\frac{1\;\text{kJ}}{1000\;\text{J}}[/latex]
[latex]{\Delta}G^{\circ} = (90.46\;-\;32.01)\;\text{kJ} = \boxed{+58.45\;\text{kJ}}[/latex]
(b) Using free energies of formation, Equation 2.
[latex]{\Delta}G_{298}^{\circ} = {\sum}n\times G_{\text{f, }298}^{\circ}(\text{products})\;-\;{\sum}n\times G_{\text{f, }298}^{\circ}(\text{reactants})[/latex]
[latex]= [1{\Delta}G_{298}^{\circ}\text{Hg}(l)\;+\;\frac{1}{2}{\Delta}G_{298}^{\circ}\text{O}_2(g)]\;-\;1{\Delta}G_{298}^{\circ}\text{HgO}(s\text{, yellow})[/latex]
[latex]= [1\;\text{mol}(0\;\text{kJ/mol})\;+\;\frac{1}{2}\text{mol}(0\;\text{kJ/mol})]\;-\;1\;\text{mol}(-58.43\;\text{kJ/mol}) = \boxed{+58.43\;\text{kJ}}[/latex]
Both ways to calculate the standard free energy change at 25 °C give the same numerical value (to three significant figures), and both predict that the process is nonspontaneous (not spontaneous) at room temperature.
Temperature-Dependence of Spontaneity
In Section 5.5, we saw that the spontaneity of a process may depend upon the temperature of the system. Phase transitions, for example, will proceed spontaneously in one direction or the other depending upon the temperature of the substance in question. Likewise, some chemical reactions can also exhibit temperature dependent spontaneities. To illustrate this concept, the equation relating free energy change to the enthalpy and entropy changes for the process is considered
[latex]{\Delta}G = {\Delta}H\;-\;T{\Delta}S[/latex]
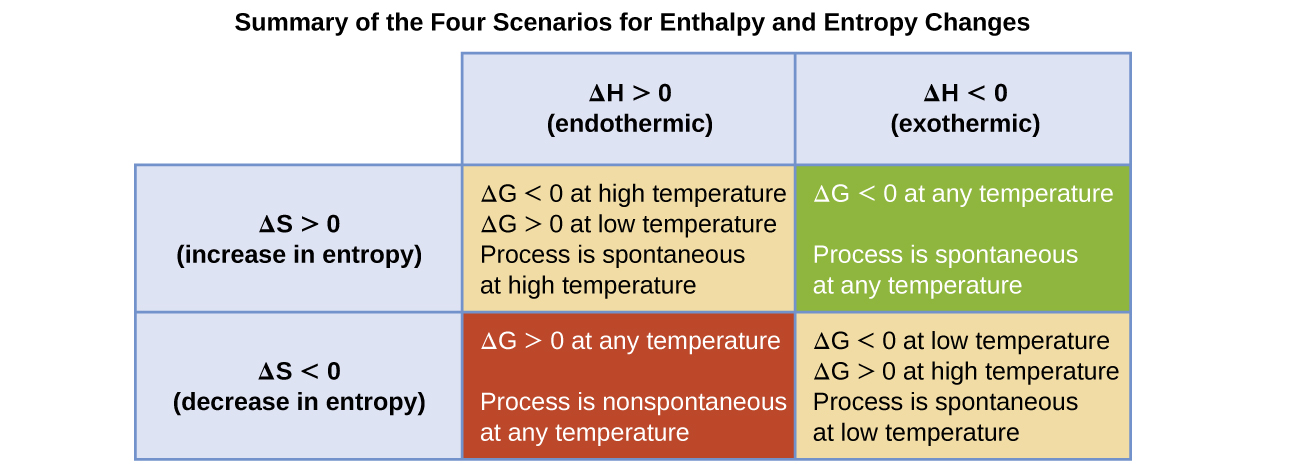
The spontaneity of a process, as reflected in the arithmetic sign of its free energy change, is then determined by the signs of the enthalpy and entropy changes and, in some cases, the absolute temperature. Since T is the absolute (kelvin) temperature, it can only have positive values. Four possibilities therefore exist with regard to the signs of the enthalpy and entropy changes:
- Both ΔH and ΔS are positive. This condition describes an endothermic process that involves an increase in system entropy. In this case, ΔG will be negative if the magnitude of the TΔS term is greater than ΔH. If the TΔS term is less than ΔH, the free energy change will be positive. Such a process is spontaneous at high temperatures and nonspontaneous at low temperatures.
- Both ΔH and ΔS are negative. This condition describes an exothermic process that involves a decrease in system entropy. In this case, ΔG will be negative if the magnitude of the TΔS term is less than ΔH. If the TΔS term’s magnitude is greater than ΔH, the free energy change will be positive. Such a process is spontaneous at low temperatures and nonspontaneous at high temperatures.
- ΔH is positive and ΔS is negative. This condition describes an endothermic process that involves a decrease in system entropy. In this case, ΔG will be positive regardless of the temperature. Such a process is nonspontaneous at all temperatures.
- ΔH is negative and ΔS is positive. This condition describes an exothermic process that involves an increase in system entropy. In this case, ΔG will be negative regardless of the temperature. Such a process is spontaneous at all temperatures.
These four scenarios are summarized in Figure 1.
Example 3
Predicting the Temperature Dependence of Spontaneity
The incomplete combustion of carbon is described by the following equation
[latex]2\text{C}(s)\;+\;\text{O}_2(g)\;{\longrightarrow}\;2\text{CO}(g)[/latex]
How does the spontaneity of this process depend upon temperature?
SOLUTION
Combustion processes are exothermic (ΔH < 0). This particular reaction involves an increase in entropy due to the accompanying increase in the amount of gaseous species (net gain of one mole of gas, ΔS > 0). The reaction is therefore spontaneous (ΔG < 0) at all temperatures.
Example 4
Equilibrium Temperature for a Phase Transition
The boiling point of a liquid is the temperature at which its solid and liquid phases are in equilibrium (that is, when vaporization and condensation occur at equal rates). Use the information (Source: OpenStax Chemistry 2e) to estimate the boiling point of water. Recall from Table 1 that ΔG = 0 at equilibrium.
SOLUTION
The process of interest is the following phase change
[latex]\text{H}_2\text{O}(l)\;{\longrightarrow}\;\text{H}_2\text{O}(g)[/latex]
When this process is at equilibrium, ΔG = 0, so the following is true
[latex]0 = {\Delta}H^{\circ}\;-\;T{\Delta}S^{\circ}\;\;\;\;\;\;\;\text{or}\;\;\;\;\;\;\;T = \frac{{\Delta}H^{\circ}}{{\Delta}S^{\circ}}[/latex]
Using the standard thermodynamic data (Source: OpenStax Chemistry 2e),
[latex]\begin{array}{r @{{}={}} l} {\Delta}H_{\text{vap}}^{\circ} &= {\Delta}H_{\text{f}}^{\circ}(\text{H}_2\text{O}(g))\;-\;{\Delta}H_{\text{f}}^{\circ}(\text{H}_2\text{O}(l)) \\[0.5em] &= -241.82\;\text{kJ/mol}\;-\;(-285.83\;\text{kJ/mol}) = 44.01\;\text{kJ/mol} \end{array}[/latex]
[latex]\begin{array}{r @{{}={}} l} {\Delta}S_{\text{vap}}^{\circ} &= {\Delta}S_{298}^{\circ}(\text{H}_2\text{O}(g))\;-\;{\Delta}S_{298}^{\circ}(\text{H}_2\text{O}(l)) \\[0.5em] &= 188.8\;\text{J/K}{\cdot}\text{mol}\;-\;70.0\;\text{J/K}{\cdot}\text{mol} = 118.8\;\text{J/K}{\cdot}\text{mol} \end{array}[/latex]
[latex]T = \frac{{\Delta}H_{\text{vap}}^{\circ}}{{\Delta}S_{\text{vap}}^{\circ}} = \frac{44.01\;\times\;10^3\;\text{J/mol}}{118.8\;\text{J/K}{\cdot}\text{mol}} = \boxed{370.5\;\text{K}} = 97.3\;^{\circ}\text{C}[/latex]
The accepted value for water’s normal boiling point is 373.2 K (100.0 °C), and so this calculation is in reasonable agreement. Note that the values for enthalpy and entropy changes data used were derived from standard data at 298 K. If desired, we could obtain more accurate results by using enthalpy and entropy changes determined at (or at least closer to) the actual boiling point.
Free Energy and Equilibrium
The free energy change for a process may be viewed as a measure of its driving force. A negative value for ΔG represents a finite driving force for the process in the forward direction, while a positive value represents a driving force for the process in the reverse direction. When ΔG is zero, the forward and reverse driving forces are equal, and so the process occurs in both directions at the same rate (the system is at equilibrium).
In Section 3.3, the reaction quotient, Q, was introduced as a convenient measure of the status of an equilibrium system. Recall that Q is the numerical value of the mass action expression for the system, and that we may use its value to identify the direction in which a reaction will proceed in order to achieve equilibrium. When Q is lesser than the equilibrium constant, K, the reaction will proceed in the forward direction until equilibrium is reached and Q = K. Conversely, if Q < K, the process will proceed in the reverse direction until equilibrium is achieved.
The free energy change for a process taking place with reactants and products present under nonstandard conditions, ΔGr, is related to the standard free energy change, ΔGr°, according to this equation:
Equation 3
[latex]{\Delta}G_{\text{r}} = {\Delta}G_{\text{r}}^{\circ}\;+\;RT\;\text{ln}\;Q[/latex]
- [latex]\Delta G_{\text{r}}[/latex], molar Gibbs free energy change of reaction ([latex]\text{J}/\text{mol}[/latex]); [latex]^\circ[/latex] denotes standard condition
- [latex]R[/latex], gas constant ([latex]8.314\;\frac{\text{J}}{\text{mol}\cdot \text{K}}[/latex])
- [latex]T[/latex], kelvin or absolute temperature
- [latex]Q[/latex], mixed reaction quotient
Previously, we defined the reaction quotients and equilibrium constants as purely in terms of concentrations (Qc or Kc) or partial pressures (Qp or Kp). Mixed reaction quotients and mixed equilibrium constants have gaseous species in partial pressures (atm or bar) and aqueous solutes in molar concentrations.
Example 5
Calculating ΔGr under Nonstandard Conditions
What is the free energy change for the process shown here under the specified conditions?
[latex]T = 25 \text{ °C}[/latex], [latex]P_{\text{N}_2} = 0.870\;\text{atm}[/latex], [latex]P_{\text{H}_2} = 0.250\;\text{atm}[/latex], and [latex]P_{\text{NH}_3} = 12.9\;\text{atm}[/latex]
[latex]2\text{NH}_3(g)\;{\longrightarrow}\;3\text{H}_2(g)\;+\;\text{N}_2(g)\;\;\;\;\;\;\;{\Delta}G_{\text{r}}^{\circ} = 33.0\;\text{kJ/mol}[/latex]
SOLUTION
Equation 3 relating free energy change to standard free energy change and reaction quotient may be used directly:
[latex]{\Delta}G_{\text{r}} = {\Delta}G_{\text{r}}^{\circ}\;+\;RT\;\text{ln}\;Q = 33.0\;\frac{\text{kJ}}{\text{mol}}\;+\;(8.314\;\frac{\text{J}}{\text{mol K}}\;\times\;298\;\text{K}\;\times\;\text{ln}\;\frac{(0.250^3)\;\times\;0.870}{12.9^2}) = \boxed{+9680\;\frac{\text{J}}{\text{mol}}}\;\text{or}\;+9.68\;\text{kJ/mol}[/latex]
Since the computed value for ΔGr is positive, the reaction is nonspontaneous under these conditions.
For a system at equilibrium, Q = K and ΔGr = 0, and Equation 3 may be written as
[latex]0 = {\Delta}G_{\text{r}}^{\circ}\;+\;RT\;\text{ln}\;K\;\;\;\;\;\;\;(\text{at equilibrium})[/latex]
and rearranged into
Equation 4
[latex]{\Delta}G_{\text{r}}^{\circ} = -RT\;\text{ln}\;K\;\;\;\;\;\;\;\text{or}\;\;\;\;\;\;\;K = e^{-\frac{{\Delta}G_{\text{r}}^{\circ}}{RT}}[/latex]
- [latex]\Delta G_{\text{r}}^\circ[/latex], standard molar Gibbs free energy of reaction ([latex]\text{J}/\text{mol}[/latex])
- [latex]R[/latex], gas constant ([latex]8.314\;\frac{\text{J}}{\text{mol}\cdot \text{K}}[/latex])
- [latex]T[/latex], kelvin or absolute temperature
- [latex]K[/latex], mixed equilibrium constant
This form of the equation provides a useful link between these two essential thermodynamic properties, and it can be used to derive equilibrium constants from standard free energy changes and vice versa. The relations between standard free energy changes and equilibrium constants are summarized in Table 2.
|
Equilibrium Constant |
Standard Molar Free Energy Change |
Comments |
|
K > 1 |
ΔG°r < 0 |
Products are more abundant at equilibrium |
|
K < 1 |
ΔG°r > 0 |
Reactants are more abundant at equilibrium |
|
K = 1 |
ΔG°r = 0 |
Reactants and products are equally abundant at equilibrium |
|
Table 2 Relations between Equilibrium Constants and Standard Free Energy Changes. Source: OpenStax Chemistry 2e |
||
Example 6
Calculating an Equilibrium Constant using Standard Free Energy Change
Given that the standard free energies of formation of Ag+(aq), Cl−(aq), and AgCl(s) are 77.1 kJ/mol, −131.2 kJ/mol, and −109.8 kJ/mol, respectively, calculate the solubility product, Ksp, for AgCl.
SOLUTION
The reaction of interest is the following
[latex]\text{AgCl}(s)\;{\rightleftharpoons}\;\text{Ag}^{+}(aq)\;+\;\text{Cl}^{-}(aq)\;\;\;\;\;\;\;K_{\text{sp}} = [\text{Ag}^{+}][\text{Cl}^{-}][/latex]
The standard free energy change for this reaction is first computed using standard free energies of formation for its reactants and products
[latex]{\Delta}G_{\text{r}}^{\circ} = [{\Delta}G_{\text{f}}^{\circ}\;(\text{Ag}^{+}(aq))\;+\;{\Delta}G_{\text{f}}^{\circ}\;(\text{Cl}^{-}(aq))]\;-\;[{\Delta}G_{\text{f}}^{\circ}\;(\text{AgCl}(s))] = [77.1\;\text{kJ/mol}\;-\;131.2\;\text{kJ/mol}]\;-\;[-109.8\;\text{kJ/mol}] = 55.7\;\text{kJ/mol}[/latex]
The equilibrium constant for the reaction may then be derived from its standard molar free energy change
[latex]K_{\text{sp}} = e^{-\frac{{\Delta}G_{\text{r}}^{\circ}}{RT}} = \text{exp}\;(-\frac{{\Delta}G_{\text{r}}^{\circ}}{RT}) = \text{exp}(-\frac{55.7\;\times\;10^3\text{J/mol}}{8.314\;\text{J/mol}{\cdot}\text{K}\;\times\;298.15\;\text{K}}) = \text{exp}(-22.470) = e^{-22.470} = \boxed{1.7\;\times\;10^{-10}}[/latex]
To further illustrate the relation between these two essential thermodynamic concepts, consider the observation that reactions spontaneously proceed in a direction that ultimately establishes equilibrium. As may be shown by plotting the free energy change versus the extent of the reaction (for example, as reflected in the value of Q), equilibrium is established when the system’s free energy is minimized, Figure 2. If a system is present with reactants and products present in nonequilibrium amounts (Q ≠ K), the reaction will proceed spontaneously in the direction necessary to establish equilibrium.
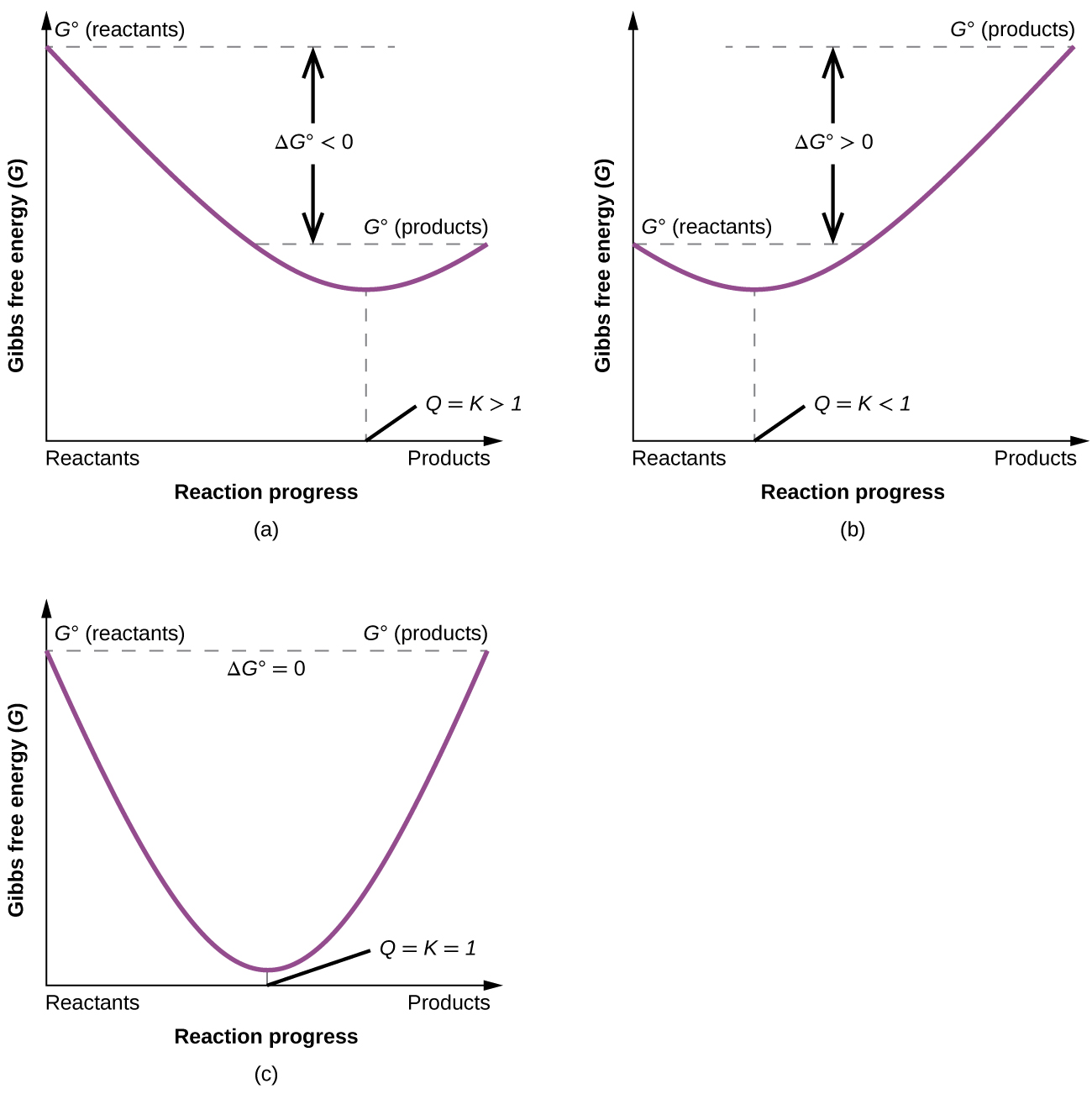
The nonzero slopes of tangent lines of the curves in Figure 2 represent nonstandard ΔG values. The slope begins with a very large negative value (extent of reaction = 0, Q = 0, ΔG is very negative). At equilibrium, slope = 0 and thus, ΔG = 0 (free energy has been minimized). Near the end, the slope has a very large positive value (extent of reaction approaches 1, Q is very large, ΔG is very positive, nonspontaneous forward reaction).
Data Table
Standard thermodynamic properties (ΔH°f, ΔG°f, and S°) for select substances (Source: OpenStax Chemistry 2e)
Optional Activity
Perform a virtual lab about the spontaneity of phase changes.
Given that the standard Gibbs free energy change is −199.7 kJ for the reaction
FeS(s) + O2(g) [latex]\longrightarrow[/latex] Fe(s) + SO2(g)
what is the standard entropy change and the value of the equilibrium constant at 298 K? The standard enthalpy of formation for FeS(s) is −100.0 kJ/mol and for SO2(g) is 296.83 kJ/mol.
Click to see answer
[latex]\begin{eqnarray} {\Delta}H^\circ &=& {\sum}n\times \Delta H^\circ_{\text{products}}\;-\;{\sum}n\times \Delta H^\circ_{\text{reactants}} \\ &=&\left[(1 \text{ mol})\left(0 \text{ kJ/mol}\right) + (1 \text{ mol})\left(296.83 \frac{\text{ kJ}}{\text{mol}}\right)\right] - \left[(1 \text{ mol})\left(-100.0 \frac{\text{ kJ}}{\text{mol}}\right) + (1 \text{ mol})\left(0 \text{ kJ/mol}\right)\right] \\ &=&396.8 \text{ kJ} \end{eqnarray}[/latex]
[latex]\begin{eqnarray} \Delta G^\circ &=& \Delta H^\circ – T \Delta S^\circ \\ \Delta S^\circ &=& \frac{\Delta H^\circ - \Delta G^\circ}{T} \\ &=& \frac{396.8 \text{ kJ} – (-199.7 \text{ kJ})}{298 K} = \boxed{2.00 \text{ kJ/K}} \end{eqnarray}[/latex]
[latex]\begin{eqnarray} \Delta G_{\text{r}}^\circ &=& – RT \ln K \\ K &=& e^{\frac{\Delta G_{\text{r}}^\circ}{–RT}} \\ &=& e^{\frac{−199.7 \times 10^3 \text{ J/mol}}{–(8.314 \text{ J/(mol K))}(298 \text{ K})}} \\ &=& \boxed{1.0 \times 10^{35}} \end{eqnarray}[/latex]
✩ UNIT 5 ASSIGNMENT
Complete Unit 5 Assignment (graded) on Moodle. This assignment is worth 11% of the course grade.
